
Background: TH17 discussed previously
here; previous brief run-down of T-cell development
here.
TH17 cells are an inflammatory T-cell subtype implicated in acute adaptive immune response as well as chronic autoimmune diseases. We have known for a while now how to create TH17 cells in a dish: just add TGFb and IL-6 (a general T-cell proliferation cytokine such as IL-2 wouldn't hurt either). However, we haven't been very clear on how exactly those kind of conditions would arise in vivo. TGFb is anti-inflammatory and helps slow the adaptive immune response down while IL-6 is pro-inflammatory and revs up the immune system. So how would these 2 molecules be made at the same time? Logically speaking, they wouldn't as it is tempting to think of the immune system as monolithic: capable of one state at a time, either ramping up inflammation or cooling it down. But because TH17 cells have been found in vivo infiltrating tissues and being made in the lymph nodes, and because we haven't yet found any other combination of cytokines that leads to the thorough development of TH17, TGFb and IL-6 must be co-expressed under some condition, somehow.
____________________________________________________________________
Before we get there, I'm going to blather at ya'll about dendritic cells for a moment (although I wish I BlogSpot had the functionality to embed this in a text-wrapping box). Dendritic cells (DCs) are the interface between the innate and adaptive immune systems. DCs are phagocytic, meaning they can gobble up pretty much any cell that they see fit to do so to. When they gobble something up, they put it into a special vacuole and crank it through molecular blenders (called proteasomes) to generate short little peptides that they then port back out to their surfaces on major histocompatibility II proteins (MHCII) that T-cells can bind to and recognize. T-cells can't recognize peptides, called antigens here, without the context of MHCII. Correct peptide-MHCII-CD4-TCR signalling tells the T-cell to do adaptive immune stuff that helps the body fight off disease.
 Figure A: A dendritic cell grown in vitro on collagen. Note the tentacley things protruding all over the place, those are the dendritic processes. Not all of the dendritic processes in this picture are from that one cell as other dendritic cell bodies are off screen. Picture somewhat altered for better contrast from original Wikimedia Commons jpeg.
Figure A: A dendritic cell grown in vitro on collagen. Note the tentacley things protruding all over the place, those are the dendritic processes. Not all of the dendritic processes in this picture are from that one cell as other dendritic cell bodies are off screen. Picture somewhat altered for better contrast from original Wikimedia Commons jpeg.
DCs also possess Toll-like receptors that recognize PAMPs (pathogen-associated molecular patterns). PAMPs include dsRNA (TLR3), flagellin (TLR5), single-stranded RNA (TLR7), unmethylated CpG DNA (TLR9) and, most importantly here, LPS (TLR4) [LPS is a constitutive component of Gram-negative bacteria, which shed it. It is also known as endotoxin.]. When a TLR binds its ligand, the DC gets activated and migrates to the lymph node where the naive T-cells are hanging out. Once there it secretes various effector cytokines that have specific actions on the T-cells. These cytokines include IL-2, IL-4, IL-6, IL-12, TGFb, TNFa, and a whole other mess of alphanumeric soup noodles.
____________________________________________________________________
Anyway, back to the conundrum of how inflammatory IL-6 and anti-inflammatory TGFb get co-expressed and make TH17 cells in vivo. It turns out that infected apoptotic cells can lead to the right co-expression. DC processing of apoptotic cells induces TGFb to throttle down inflammation in case the immune system has gotten too aggressive and is killing off the host organism (as occurs in septic shock). DC processing of non-host infectious agents usually induces IL-6. So when a DC processes an apoptotic cell that was infected with an intracellular microbe of some sort, both stimuli are right there and the DC trundles off to the lymph node making both TGFb and IL-6.
The group in the paper below used LPS-loaded apoptotic B-cells and E. coli-infected apoptotic neutrophils to test this out. They mixed the B-cells and neutrophils with DCs and let them stew for a while as the DC chewed thoughtfully and decided what to do. They then collected the culture medium off fo the DCs, which would contain any differentiation and/or signalling cytokines the DCs secreted in response to the apoptotic cell stimulus. Next, this medium was added to cultures of naive CD4+ T-cells and they sat back and watched what happened via flow cytometry, mRNA expression, and production of phenotypic marker cytokines (specifically, IL-10 and IL-17).
Using unloaded apoptotic B-cells or uninfected apoptotic neutrophils didn't do much by way of making TH17 cells as only TGFb was produced (made Tregs, though, see below). However, this was reversible through complementation with exogenous IL-6. Also, DCs exposed just to LPS such that TLR4 was strongly activated did induce lots of IL-6 production, but this didn't lead to much TH17 production unless IFNg was blocked (this consequently blocks the production of TH1 cells).
It turns out that apoptotic cells also induce a Treg response through TGFb. Tregs are the cells that calm the rest of the immune system down when it's gotten too excited. They are characterized by expression of Fox3p and secretion of IL-10. A sizeable portion of the naive T-cells also made IL-10 and flow cytometric analysis revealed that some had become double T-cells, secreting both IL-10 and IL-17. However, this was only during the initial stimulation of the naive T-cells. Later on, after these now-differentiated T-cells had calmed down, re-stimulation with IL-23 increased IL-17 production and downregulated IL-10 mRNA, indicating that this dual expansion may be a transient phenomenon.
The group replicated their findings in vivo. They used a Citrobacter rodentium model of hemorrhagic colitis in which many cells in the gastrointestinal epithelium go apoptotic and inhibited that apoptosis. Inhibited apoptosis led to lower infiltration of TH17 cells than untreated mice. There were also a bunch of good genetic and chemical controls, but I'm not going to discuss all of that here because 1) it's a LOT of detail and 2) I'm more interested in blathering about the implications of this finding.
Implication 1: Ulcerative colitis
The sick gut cells in ulcerative colitis aren't necessarily infected themselves. But due to the very high native colonization of the gastrointestinal tract by our friendly microbiota, there are always relatively high levels of LPS in the lumen that, presumably, interact with the epithelium. Therefore, at the ulcers, especially those mediated by Helicobacter spp., there is an increased likelihood that responding DCs will encounter both microbial PAMPs and apoptotic cells. This could explain, at least partly, why TH17 cells are heavily involved in the inflammation associated with gastritis.
Implication 2: Cancer
Given that pre-cancerous cells are generally not listening to the cells around them and have higher rates of mutation, it is entirely plausable that some cancer cells with alter the expression patterns of MHCI on their surface, be recognized by CD8+ T-cells, and summarily executed. However, were a DC to encounter this apoptotic body in the abscence of a TLR ligand, it would go to the lymph node promoting development of Tregs for the cancer cell antigens*. While this may be a good thing in preventing autoimmune reactions when T-cells learn the unmutated antigens still present in cancer cells are bad and should be killed, it also brings up the somewhat unsettling prospect that the this leads to immunotolerance of cancerous cells. The immune system is generally the first line of defense against the development of tumors, and if it has specifically learned to not be reactive against them due to Tregs, then the cancer would be allowed to continue growing and mutating without immune system interference. Needless to say, this is ultimately bad for the organism.
Implication 3: Rheumatoid Arthritis
It is known that inflammed tissues produce IL-23, which encourages stable differentiation of TH17 cells and also acts as a chemoattractant for them. I don't think it's unreasonable to think that levels of IL-23, along with NFkB et al, would be elevated in arthritic tissues and that this could lead to infiltration of TH17 cells. Once there, TH17 cells can cause further inflammation, including tissue damage. This in turn would amplify the production of IL-23, which has been shown to decrease IL-10 production and in turn decrease Tregs. This then leads to a dismal scenario in which more and more TH17 cells are being recruited to drive inflammation in arthritic tissue while at the same time stomping on the Tregs that could help break that feedback loop. However, at the same time DCs would be present and processing the apoptotic cells in the arthritic tissues, and then hopefully driving towards TGFb and Tregs.
It should be noted that each and every implication I have put up above could be complete and utter horsefeathers as the immune system is so complex and operates on so many scales that I could very easily be overlooking a key component that makes all of my conjectures seem silly.
Torchinsky, M., Garaude, J., Martin, A., & Blander, J. (2009). Innate immune recognition of infected apoptotic cells directs TH17 cell differentiation Nature, 458 (7234), 78-82 DOI: 10.1038/nature07781
*This happens anyway, MHCI or not. B7.1 and B7.2 (CD80 and CD86, respectively) expression patterns are often altered in chronic myelogenous leukemia and chorionic gonadotropic, which isn't expressed in adults, sometimes pops back up in tumors.

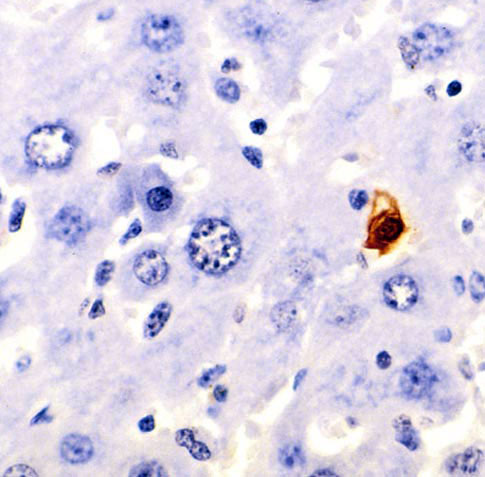 Figure A: TUNEL histochemical staining in murine liver, brown cell is apoptotic.
Figure A: TUNEL histochemical staining in murine liver, brown cell is apoptotic.




